rotl tutorial
for v3.0.3
rotl provides an interface to the Open Tree of Life (OTL) API and allows users
to query the API, retrieve parts of the Tree of Life and integrate these parts
with other R packages.
The OTL API provides services to access:
- the Tree of Life a.k.a. TOL (the synthetic tree): a single draft tree that is a combination of the OTL taxonomy and the source trees (studies)
- the Taxonomic name resolution services a.k.a. TNRS: the methods for
resolving taxonomic names to the internal identifiers used by the TOL and the
GOL (the
ott ids). - the Taxonomy a.k.a. OTT (for Open Tree Taxonomy): which represents the synthesis of the different taxonomies used as a backbone of the TOL when no studies are available.
- the Studies containing the source trees used to build the TOL, and extracted from the scientific literature.
In rotl, each of these services correspond to functions with different
prefixes:
| Service | rotl prefix |
|---|---|
| Tree of Life | tol_ |
| TNRS | tnrs_ |
| Taxonomy | taxonomy_ |
| Studies | studies_ |
rotl also provides a few other functions and methods that can be used to
extract relevant information from the objects returned by these functions.
Installation
install.packages("rotl")
Or development version from GitHub
install.packages("devtools")
devtools::install_github("ropensci/rotl")
library("rotl")
Usage
Demonstration of a basic workflow
The most common use for rotl is probably to start from a list of species and
get the relevant parts of the tree for these species. This is a two step
process:
- the species names need to be matched to their
ott_id(the Open Tree Taxonomy identifiers) using the Taxonomic name resolution services (TNRS) - these
ott_idwill then be used to retrieve the relevant parts of the Tree of Life.
Step 1: Matching taxonomy to the ott_id
Let’s start by doing a search on a diverse group of taxa: a tree frog (genus Hyla), a fish (genus Salmo), a sea urchin (genus Diadema), and a nautilus (genus Nautilus).
taxa <- c("Hyla", "Salmo", "Diadema", "Nautilus")
resolved_names <- tnrs_match_names(taxa)
It’s always a good idea to check that the resolved names match what you intended:
| search_string | unique_name | approximate_match | ott_id | is_synonym | flags | number_matches |
|---|---|---|---|---|---|---|
| hyla | Hyla | FALSE | 1062216 | FALSE | 1 | |
| salmo | Salmo | FALSE | 982359 | FALSE | 1 | |
| diadema | Diadema (genus in Nucletmycea) | FALSE | 4930522 | FALSE | 5 | |
| nautilus | Nautilus | FALSE | 616358 | FALSE | 1 |
The column unique_name sometimes indicates the higher taxonomic level
associated with the name. The column number_matches indicates the number of
ott_id that corresponds to a given name. In this example, our search on
Diadema returns 2 matches, and the one returned by default is indeed the sea
urchin that we want for our query. The argument context_name allows you to
limit the taxonomic scope of your search. Diadema is also the genus name of a
fungus. To ensure that our search is limited to animal names, we could do:
resolved_names <- tnrs_match_names(taxa, context_name = "Animals")
If you are trying to build a tree with deeply divergent taxa that the argument
context_name cannot fix, see “How to change the ott ids assigned to my taxa?”
in the FAQ below.
Step 2: Getting the tree corresponding to our taxa
Now that we have the correct ott_id for our taxa, we can ask for the tree
using the tol_induced_subtree() function. By default, the object returned by
tol_induced_subtree is a phylo object (from the
ape package), so we can plot it
directly.
my_tree <- tol_induced_subtree(ott_ids = resolved_names$ott_id)
plot(my_tree, no.margin=TRUE)
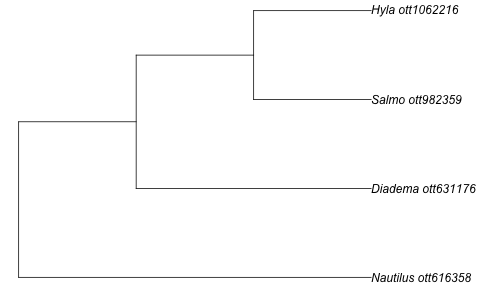
Get tree for a particular taxonomic group
If you are looking to get the tree for a particular taxonomic group, you need to
first identify it by its node id or ott id, and then use the tol_subtree()
function:
mono_id <- tnrs_match_names("Monotremes")
mono_tree <- tol_subtree(ott_id = mono_id$ott_id[1])
plot(mono_tree)

Find trees from studies focused on my favourite taxa
The function studies_find_trees() allows the user to search for studies
matching a specific criteria. The function studies_properties() returns the
list of properties that can be used in the search.
furry_studies <- studies_find_studies(property="ot:focalCladeOTTTaxonName", value="Mammalia")
furry_ids <- furry_studies$study_ids
Now that we know the study_id, we can ask for the meta data information
associated with this study:
furry_meta <- get_study_meta("pg_2550")
get_publication(furry_meta) ## The citation for the source of the study
#> [1] "O'Leary, Maureen A., Marc Allard, Michael J. Novacek, Jin Meng, and John Gatesy. 2004. \"Building the mammalian sector of the tree of life: Combining different data and a discussion of divergence times for placental mammals.\" In: Cracraft J., & Donoghue M., eds. Assembling the Tree of Life. pp. 490-516. Oxford, United Kingdom, Oxford University Press."
#> attr(,"DOI")
#> [1] ""
get_tree_ids(furry_meta) ## This study has 10 trees associated with it
#> [1] "tree5513" "tree5515" "tree5516" "tree5517" "tree5518" "tree5519"
#> [7] "tree5520" "tree5521" "tree5522" "tree5523"
candidate_for_synth(furry_meta) ## None of these trees are yet included in the OTL
#> NULL
Using get_study("pg_2550") would returns a multiPhylo object (default) with
all the trees associated with this particular study, while
get_study_tree("pg_2550", "tree5513") would return one of these trees.
Citing
To cite rotl in publications use:
Michonneau, F., Brown, J. W. and Winter, D. J. (2016), rotl: an R package to interact with the Open Tree of Life data. Methods Ecol Evol. 7(12):1476-1481. doi:10.1111/2041-210X.12593
License and bugs
- License: BSD_2_clause
- Report bugs at our Github repo for rotl
